Dietary Salt Impairs the Endothelial Glycocalyx: The Most Important Cardiovascular Disease Risk Factor You May Never Have Heard About
Frequently, important physiological discoveries made by scientists who study obscure topics escape the attention of the general public, health professionals, and even other scientists. Such has been the case for the endothelial glycocalyx: a delicate and fragile structure lining the inside surface of all blood vessels.
The endothelial glycocalyx had been inferred from blood flow measurements as far back as the 1940’s, but due to its fragile configuration the structure had never been viewed until 1966 when it was first detected with an electron microscope using special staining procedures (1). Over the next 30 years, scientists speculated about the function of the endothelial glycocalyx, but it was not until 1996 that two researchers, Vink and Duling (2), provided an answer. They concluded that the endothelial glycocalyx “may represent the true active interface between blood and the capillary wall”.
Okay, no big deal. A tiny and fragile blood vessel structure was first visualized 50 years ago and then it took scientists another 30 years to assign what many would consider a minor function to it. But is the function of the endothelial glycocalyx really that minor?
In fact, the glycocalyx is so important that its dysfunction plays a critical role in almost every step of atherosclerosis – the process that leads to heart disease and stroke. And as you’ll see, dietary sodium can directly contribute to this dysfunction and start the disease process.
Visualizing the Glycocalyx
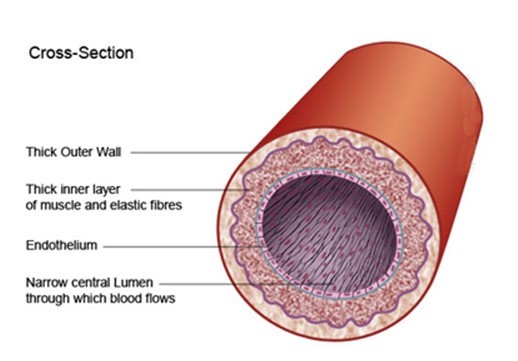
I tend to be a visual learner, as do many people. So, let’s take a look at the cross section of an artery, the way many cardiologists visualized it prior to 1996. Figure 1 below shows that the inner-most surface of arteries is lined by cells called the endothelium. However, nowhere in sight is the endothelial glycocalyx. This perception of blood vessel structure was commonly held until about 1994 when a select group of scientists developed a new staining procedure (alcian blue) to better visualize the endothelial glycocalyx (1).
Figure 1. Classical visualization of an artery cross section
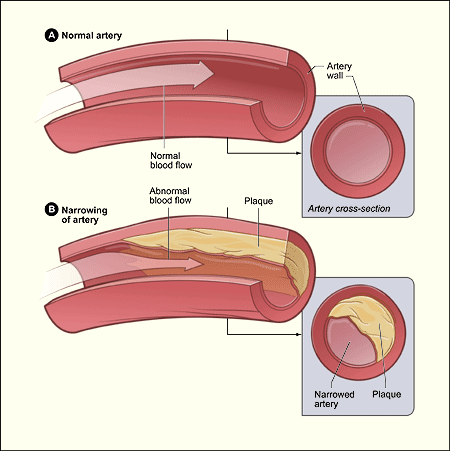
The widespread impression of blood vessels devoid of an endothelial glycocalyx gave scientists studying cardiovascular disease an imperfect view of the atherosclerotic process which underlies heart attacks and strokes. Figure 2 shows the commonly held perspective of the day for atherosclerosis in a blood vessel without an endothelial glycocalyx.
Figure 2. Classical visualization of atherosclerosis without the endothelial glycocalyx.
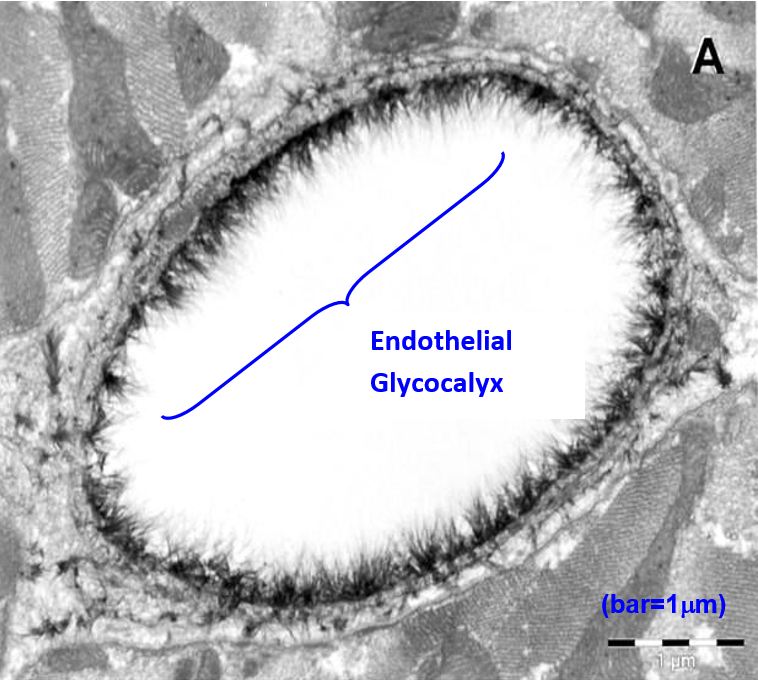
In contrast, Figure 3 shows the cross section of a capillary taken in 2003 using electron microscopy with alcian blue staining procedures. It provides indisputable evidence of the endothelial glycocalyx (3).
Figure 3. Electron microscope overview of an alcian blue stained rat left ventricular myocardial capillary showing the endothelial glycocalyx (3).
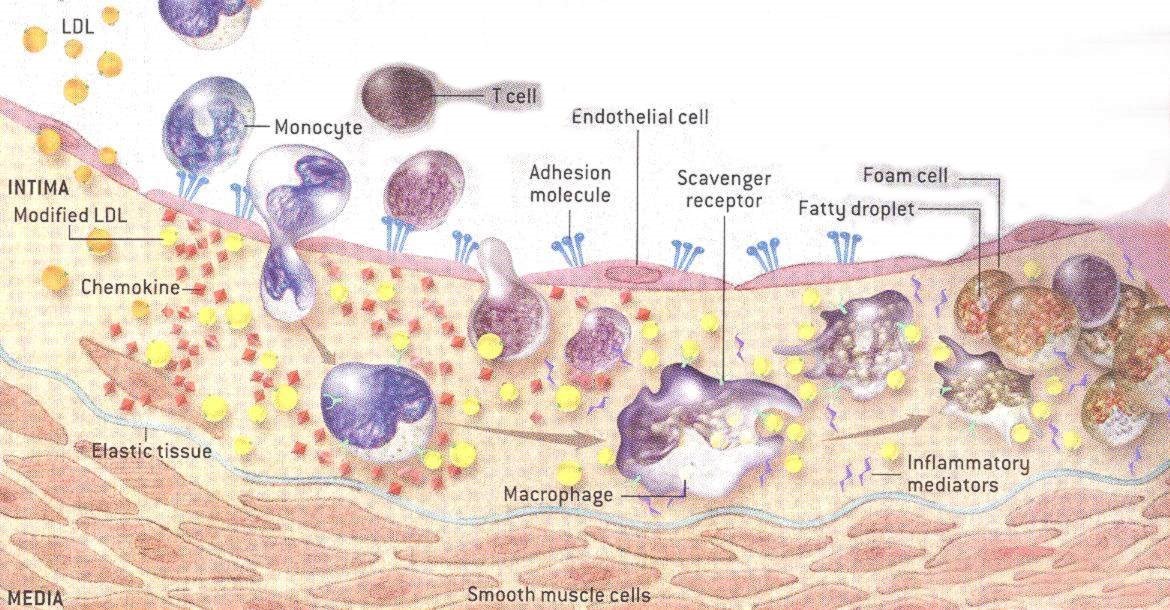
When an atherosclerotic plaque forms in the intima of arteries, a number of key steps occur which can ultimately lead to cardiovascular disease: 1) Inflammation (8, 14, 19, 77) causes the endothelial glycocalyx to be shed (14-29), promoting further inflammation. 2) The glycocalyx becomes thinner in areas of turbulent blood flow (1, 7, 21, 23) where atherosclerotic plaques form. 3) The shedding of the glycocalyx promotes movement of LDL cholesterol from the bloodstream into the intima (20, 22, 28). 4) The LDL cholesterol becomes oxidized and continues to promote glycocalyx shedding. 5) Leukocytes, monocytes, T cells, other leukocytes and platelets, attach to adhesion molecules on the surface of endothelial cells (4-6, 12, 15, 17, 18, 26, 33) and then enter the intima. 6) After entry into the arterial intima, a specific type of leukocyte, called monocytes, are transformed into macrophages. 7) Macrophages consume oxidized LDL cholesterol and other elements to become foam cells. 8) Over time and continued inflammation, these foam cells become the atherosclerotic plaques that clog arteries. 9) Eventually a fibrous cap forms over the plaque which, if ruptured via the continued inflammatory processes, can precipitate heart attacks and strokes.
The atherosclerotic process is more complex than these simple nine steps, but these steps will provide you with sufficient information to understand how a high salt diet promotes atherosclerosis at nearly every step along the way.
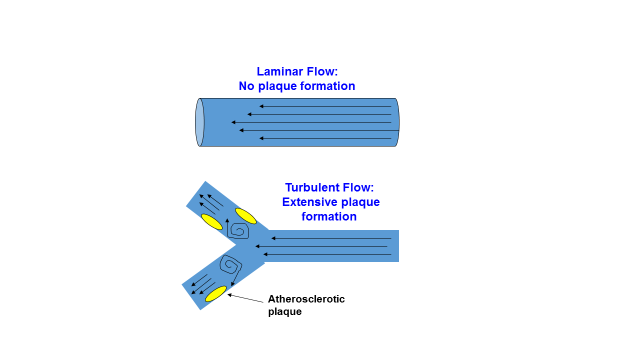
However, before I show you how a high salt diet damages the endothelial glycocalyx and promotes CVD, heart attacks and strokes, one more important detail must be known. An additional revealing clue to the atherosclerotic process is the anatomical location of atherosclerotic plaques within arteries. As far back as 1971, C.G. Caro and colleagues (52, 53) showed that early atherosclerotic lesions developed almost exclusively in areas of turbulent blood flow near bifurcations (forks) of arteries, and rarely in areas of non-turbulent (laminar) blood flow. This fact has been verified in all modern studies of atherosclerosis (1, 7, 21, 21, 23, 53). Figure 5 below shows this phenomenon of plaque formation in areas of turbulent flow where the thickness and density of the endothelial glycocalyx is reduced. Current evidence indicates that atherosclerosis arises in these areas of turbulent flow because they are less able (than areas of non-turbulent flow) to respond to inflammatory conditions in the bloodstream, which promote glycocalyx shedding (8, 14, 19, 77).
Figure 5. Effect of turbulent blood flow and laminar (non-turbulent) blood flow upon the distribution of atherosclerotic plaques in arteries.
How a High Salt Diet Promotes Atherosclerosis and Cardiovascular Disease (CVD)
Normally, plasma concentrations of sodium are maintained within a narrow range that varies in the general population from 134 to 148 mmol/L (54-56). Increasing dietary salt in subjects without hypertension elevates plasma sodium concentrations within or beyond the normal range (57, 58).
In the Paleo diet community, a common perception is that adding salt, or particularly sea salt, to a Paleo diet has little or no adverse effect upon health and wellbeing (59-68). Nevertheless, the data reveals that salt, even added to the diet of normotensives (normal blood pressure), elevates plasma sodium concentrations and has important and far-reaching health considerations; particularly if the salt habit is continued throughout life.
Experiments conducted in 2007 on living human endothelial cells incubated outside the body (ex vivo) demonstrated that stiffness of the cells was unaffected by sodium concentrations of < 135 mmol/L, but stiffness rose steeply at concentrations between 135 and 145 mmol/L (70). Further, higher plasma sodium concentrations (~143-150 mmol/L), which are still mostly within normal ranges and can easily be achieved in living humans by adding salt to their diet (54-58), caused the endothelial cells to stiffen and reduce their production of nitric oxide (70, 72): a compound found throughout the body. In blood vessels, nitric oxide suppresses cell inflammation and adhesion of leukocytes and platelets to the endothelium (71). In this way, normal nitric oxide production by arterial endothelial cells inhibits blood clotting and promotes normal blood flow. Nitric oxide metabolism and production is diminished with age and CVD (71).
One of the problems with the ex vivo experiment conducted in 2007 (70) was that the endothelial cells were not only incubated with sodium, but also with a hormone called aldosterone. Accordingly, the interpretation of the experiment was muddled. Was it the sodium, the aldosterone, or both that caused a stiffening of the endothelial cells and decreased production of nitric oxide? In the ensuing years, others researchers replicated these results (46, 72), but the question still remained: what caused the adverse effects upon endothelial function and nitric oxide production?
This question was finally answered in 2016 in a living (in vitro) mouse model that had a genetic mutation preventing it from synthesizing aldosterone [aldosterone synthase knockout mouse] (69). A high salt diet, without aldosterone, increased plasma sodium to 150 mmol/L, increased endothelial cell stiffness by 44 percent, and suppressed nitric oxide production. These results are consistent with two recent randomized controlled human trials showing that a high salt meal (65 mmol Na) increased plasma sodium concentrations and impaired endothelial function while increasing arterial stiffness (74, 75).
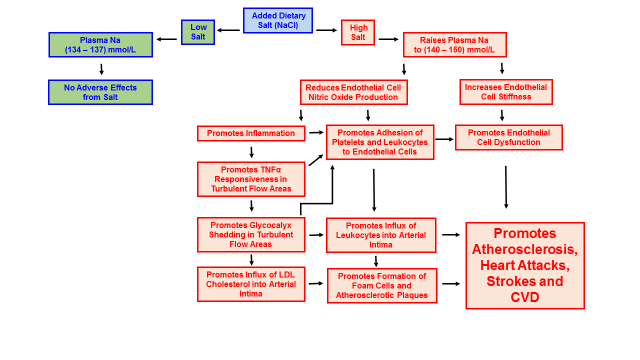
In a living (in vivo) animal model, high salt diets fed to mice increased the responsiveness of arterial endothelial cells to a pro-inflammatory cytokine (TNFα), at areas of turbulent blood flow within the arteries. High salt diets also increased adhesion of leukocytes to these same areas of turbulent flow (76). Both of these physiological changes are characteristic of, and precede the development of CVD in humans. As I previously mentioned, high salt diets promote inflammation by impairing the release of nitric oxide (69, 70, 72), while TNFα directly causes endothelial cells to shed the glycocalyx (8, 14, 19, 77). Accordingly, glycocalyx shedding initiated by the inflammation produced from a high salt diet facilitates the formation of atherosclerotic plaques and the development of CVD. Figure 6 below summarizes the effects of high salt diets upon the development of CVD.
Figure 6. How high salt diets promote the development of cardiovascular disease (CVD)
Clinical data from the Atherosclerosis Risk in Communities Study involving 12,779 subjects demonstrated that plasma sodium concentrations represent a significant predictor of 10-years risk of coronary heart disease (77). These findings show that elevation of plasma sodium even within the normal physiological range (134 to 148 mmol/L) is accompanied by cardiovascular changes that facilitate the development of CVD (77).
Practical Implications and Final Thoughts
Perhaps the most obvious implication of this recent, enormous and persuasive scientific literature, is simply not to add salt or sea salt to your already healthful, natural and fresh Paleo foods and recipes. Unadulterated, natural, fresh Paleo foods have very little normally occurring sodium. In a previous blog table, I have demonstrated how it is virtually impossible to ingest more than 2300 mg of sodium from natural, un-salted Paleo foods. From this table, also notice how the potassium content of natural, un-salted Paleo diets typically contains about five times more potassium than sodium.
High dietary levels of potassium are known to overcome many of the adverse cardiovascular effects of sodium by softening vascular endothelial cells (78), increasing nitric oxide production (78) and preventing sodium induced impairment of endothelial function (75). So, practically speaking, it may not always be possible to avoid foods with added salt. If this is the case, try to counter high salt foods with plenty of potassium rich fruits and vegetables. Some of my favorite high potassium foods are peaches, cherries, bananas, dates, raisins, dried figs, pecans, filberts, tomatoes, steamed broccoli and summer squash.
Another practical way to counter the effects of temporary overindulgence in salty foods is to drink more water. For an average person with an elevated plasma sodium concentration of 143 mmol/L, drinking one liter of water over a six to eight-hour period can reduce this value to the lower range of 137 mmol/L, providing no additional high salt foods are consumed (77).
So, the take home message to prevent salt induced CVD is this: 1) don’t put added salt or sea salt into fresh, healthy Paleo foods; 2) avoid salted, processed foods whenever possible. If you happen to overindulge in salted foods: 3) eat plenty of potassium rich fruits and vegetables and 4) drink more water.
References
1. Pries AR, Secomb TW, Gaehtgens P. The endothelial surface layer. Pflugers Arch. 2000 Sep;440(5):653-66
2. Vink H, Duling BR. Identification of distinct luminal domains for macromolecules, erythrocytes, and leukocytes within mammalian capillaries. Circ Res. 1996 Sep;79(3):581-9.
3. van den Berg BM, Vink H, Spaan JA. The endothelial glycocalyx protects against myocardial edema. Circ Res. 2003 Apr 4;92(6):592-4.
4. Alphonsus CS, Rodseth RN. The endothelial glycocalyx: a review of the vascular barrier. Anaesthesia. 2014 Jul;69(7):777-84.
5. Becker BF, Chappell D, Bruegger D, Annecke T, Jacob M. Therapeutic strategies targeting the endothelial glycocalyx: acute deficits, but great potential. Cardiovasc Res. 2010 Jul 15;87(2):300-10.
6. Becker BF, Chappell D, Jacob M. Endothelial glycocalyx and coronary vascular permeability: the fringe benefit. Basic Res Cardiol. 2010 Nov;105(6):687-701
7. Gouverneur M, Van Den Berg B, Nieuwdorp M, Stroes E & Vink H. Vasculoprotective properties of the endothelial glycocalyx: effects of fluid shear stress. Journal of Internal Medicine 2006; 259: 393–40
8. Kolářová H, Ambrůzová B, Svihálková Šindlerová L, Klinke A, Kubala L. Modulation of endothelial glycocalyx structure under inflammatory conditions. Mediators Inflamm. 2014;2014:694312. doi: 10.1155/2014/694312. Epub 2014 Apr 3
9. Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch Eur J Physiol (2007) 454:345–359
10. Reitsma S, oude Egbrink MG, Vink H, van den Berg BM, Passos VL, Engels W, Slaaf DW, van Zandvoort MA. Endothelial glycocalyx structure in the intact carotid artery: a two-photon laser scanning microscopy study. J Vasc Res. 2011;48(4):297-306.
11. Rorije N, Engberink RO, van den Born B, Verberne H, Vogt L. 7d.06: Effects of an acute and chronic salt load on microvascular permeability in healthy subjects. J Hypertens. 2015 Jun;33 Suppl 1:e101. doi: 10.1097/01.hjh.0000467623.14967.fc.
12. van den Berg BM, Nieuwdorp M, Stroes ES, Vink H. Glycocalyx and endothelial (dys) function: from mice to men. Pharmacol Rep. 2006;58 Suppl:75-80
13. Vincent PE, Weinberg PD. Flow-dependent concentration polarization and the endothelial glycocalyx layer: multi-scale aspects of arterial mass transport and their implications for atherosclerosis. Biomech Model Mechanobiol. 2014 Apr;13(2):313-26
14. Cancel LM, Ebong EE, Mensah S, Hirschberg C, Tarbell JM. Endothelial glycocalyx, apoptosis and inflammation in an atherosclerotic mouse model. Atherosclerosis. 2016 Sep;252:136-46.
15. Chappell D, Brettner F, Doerfler N, Jacob M, Rehm M, Bruegger D, Conzen P, Jacob B, Becker BF. Protection of glycocalyx decreases platelet adhesion after ischaemia/reperfusion: an animal study. Eur J Anaesthesiol. 2014 Sep;31(9):474-81.
16. Chappell D, Bruegger D, Potzel J, Jacob M, Brettner F, Vogeser M, Conzen P, Becker BF, Rehm M. Hypervolemia increases release of atrial natriuretic peptide and shedding of the endothelial glycocalyx. Crit Care. 2014 Oct 13;18(5):538. [Epub ahead of print]
17. Chappell D, Dörfler N, Jacob M, Rehm M, Welsch U, Conzen P, Becker BF. Glycocalyx protection reduces leukocyte adhesion after ischemia/reperfusion. Shock. 2010 Aug;34(2):133-9
18. Constantinescu AA, Vink H and Spaan JA. Endothelial surface endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler Thromb Vasc Biol 2003;23;1541-1547.
19. Henry, CBS, Duling, BR. TNF-a increases entry of macromolecules into luminal endothelial cell glycocalyx. Am J Physiol Heart Circ Physiol. 2000; 279: H2815–H2823.
20. Kang H, Fan Y, Sun A, Deng X. Compositional or charge density modification of the endothelial glycocalyx accelerates flow-dependent concentration polarization of low-density lipoproteins. Exp Biol Med (Maywood). 2011 Jul;236(7):800-7
21. Koo A, Dewey CF Jr, García-Cardeña G. Hemodynamic shear stress characteristic of atherosclerosis-resistant regions promotes glycocalyx formation in cultured endothelial cells. Am J Physiol Cell Physiol. 2013 Jan 15;304(2):C137-46
22. Liu X, Fan Y, Deng X. Effect of the endothelial glycocalyx layer on arterial LDL transport under normal and high pressure. J Theor Biol. 2011 Aug 21;283(1):71-81
23. Lopez-Quintero SV, Cancel LM, Pierides A, Antonetti D, Spray DC, Tarbell JM. High glucose attenuates shear-induced changes in endothelial hydraulic conductivity by degrading the glycocalyx. PLoS One. 2013 Nov 18;8(11):e78954. doi: 10.1371/journal.pone.0078954
24. Nijst P, Verbrugge FH, Grieten L, Dupont M, Steels P, Tang WH, Mullens W. The pathophysiological role of interstitial sodium in heart failure. J Am Coll Cardiol. 2015 Feb 3;65(4):378-88.
25. Noble MI, Drake-Holland AJ, Vink H. Hypothesis: arterial glycocalyx dysfunction is the first step in the atherothrombotic process. QJM. 2008 Jul;101(7):513-8
26. Reitsma S, Oude Egbrink MG, Heijnen VV, Megens RT, Engels W, Vink H, Slaaf DW, van Zandvoort MA. Endothelial glycocalyx thickness and platelet-vessel wall interactions during atherogenesis. Thromb Haemost. 2011 Nov;106(5):939-46
27. van den Berg BM, Spaan JA, Rolf TM, Vink H. Atherogenic region and diet diminish glycocalyx dimension and increase intima-to-media ratios at murine carotid artery bifurcation. Am J Physiol Heart Circ Physiol. 2006 Feb;290(2):H915-20.
28. van den Berg BM, Spaan JA, Vink H. Impaired glycocalyx barrier properties contribute to enhanced intimal low-density lipoprotein accumulation at the carotid artery bifurcation in mice. Pflugers Arch. 2009 Apr;457(6):1199-206
29. Voyvodic PL, Min D, Liu R, Williams E, Chitalia V, Dunn AK, Baker AB.
Loss of syndecan-1 induces a pro-inflammatory phenotype in endothelial cells with a dysregulated response to atheroprotective flow. J Biol Chem. 2014 Apr 4;289(14):9547-59.
30. Bruegger D, Schwartz L, Chappell D, Jacob M, Rehm M, Vogeser M, Christ F, Reichart B, Becker BF. Release of atrial natriuretic peptide precedes shedding of the endothelial glycocalyx equally in patients undergoing on- and off-pump coronary artery bypass surgery. Basic Res Cardiol. 2011 Nov;106(6):1111-21
31. Bruegger D, Jacob M, Rehm M, Loetsch M, Welsch U, Conzen P, Becker BF. Atrial natriuretic peptide induces shedding of endothelial glycocalyx in coronary vascular bed of guinea pig hearts. Am J Physiol Heart Circ Physiol. 2005 Nov;289(5):H1993-9.
32. Chappell D, Bruegger D, Potzel J, Jacob M, Brettner F, Vogeser M, Conzen P, Becker BF, Rehm M. Hypervolemia increases release of atrial natriuretic peptide and shedding of the endothelial glycocalyx. Crit Care. 2014 Oct 13;18(5):538. doi: 10.1186/s13054-014-0538-5.
33. Chappell D, Brettner F, Doerfler N, Jacob M, Rehm M, Bruegger D, Conzen P, Jacob B, Becker BF. Protection of glycocalyx decreases platelet adhesion after ischaemia/reperfusion: an animal study. Eur J Anaesthesiol. 2014 Sep;31(9):474-81
34. Dane M, van den Berg B, Rabelink T. The endothelial glycocalyx: scratching the surface for cardiovascular disease in kidney failure. Atherosclerosis. 2014 Jul;235(1):56-7. doi: 10.1016/j.atherosclerosis.2014.04.005. Epub 2014 Apr 25.
35. Kliche K, Gerth U, Pavenstädt H, Oberleithner H. Recharging red blood cell surface by hemodialysis. Cell Physiol Biochem. 2015;35(3):1107-15.
36. Korte S, Wiesinger A, Straeter AS, Peters W, Oberleithner H, Kusche-Vihrog K. Firewall function of the endothelial glycocalyx in the regulation of sodium homeostasis. Pflugers Arch. 2012 Feb;463(2):269-78.
37. Lee DH, Dane MJ, van den Berg BM, Boels MG, van Teeffelen JW, de Mutsert R, den Heijer M, Rosendaal FR, van der Vlag J, van Zonneveld AJ, Vink H, Rabelink TJ; NEO study group. Deeper penetration of erythrocytes into the endothelial glycocalyx is associated with impaired microvascular perfusion. PLoS One. 2014 May 9;9(5):e96477. doi: 0.1371/journal .pone.0096477
38. Oberleithner H, Wilhelmi M. Vascular glycocalyx sodium store – determinant of salt sensitivity? Blood Purif. 2015;39(1-3):7-10.
39. Oberleithner H. Sodium selective erythrocyte glycocalyx and salt sensitivity in man. Pflugers Arch. 2015 Jun;467(6):1319-25.
40. Oberleithner H, Wälte M, Kusche-Vihrog K. Sodium renders endothelial cells sticky for red blood cells. Front Physiol. 2015 Jun 30;6:188. doi: 10.3389/fphys.2015.00188.
41. Oberleithner H. Sodium selective erythrocyte glycocalyx and salt sensitivity in man. Pflugers Arch. 2014 Jul 17. [Epub ahead of print] PMID: 25027385
42. Oberleithner H. Vascular endothelium: a vulnerable transit zone for merciless sodium. Nephrol Dial Transplant. 2014 Feb;29(2):240-6.
43. Oberleithner H, Wilhelmi M. Determination of erythrocyte sodium sensitivity in man. Pflugers Arch. 2013 Oct;465(10):1459-66
44. Oberleithner H. Vascular endothelium leaves fingerprints on the surface of erythrocytes. Pflugers Arch. 2013 Oct;465(10):1451-8.
45. Oberleithner H. Two barriers for sodium in vascular endothelium? Ann Med. 2012 Jun;44 Suppl 1:S143-8
46. Oberleithner H, Peters W, Kusche-Vihrog K, Korte S, Schillers H, Kliche K, Oberleithner K. Salt overload damages the glycocalyx sodium barrier of vascular endothelium. Pflugers Arch. 2011 Oct;462(4):519-28.
47. Paar M, Pavenstädt H, Kusche-Vihrog K, Drüppel V, Oberleithner H, Kliche K. Endothelial sodium channels trigger endothelial salt sensitivity with aging. Hypertension. 2014 Aug;64(2):391-6.
48. Padberg JS, Wiesinger A, di Marco GS, Reuter S, Grabner A, Kentrup D, Lukasz A, Oberleithner H, Pavenstädt H, Brand M, Kümpers P. Damage of the endothelial glycocalyx in chronic kidney disease. Atherosclerosis. 2014 Jun;234(2):335-43.
49. Krähling H, Mally S, Eble JA, Noël J, Schwab A, Stock C. The glycocalyx maintains a cell surface pH nanoenvironment crucial for integrin-mediated migration of human melanoma cells. Pflugers Arch. 2009 Oct;458(6):1069-83.
50. Cai B, Fan J, Zeng M, Zhang L, Fu BM. Adhesion of malignant mammary tumor cells MDA-MB-231 to microvessel wall increases microvascular permeability via degradation of endothelial surface glycocalyx. J Appl Physiol (1985). 2012 Oct;113(7):1141-53.
51. Reitsma S, oude Egbrink MG, Vink H, van den Berg BM, Passos VL, Engels W, Slaaf DW, van Zandvoort MA. Endothelial glycocalyx structure in the intact carotid artery: a two-photon laser scanning microscopy study. J Vasc Res. 2011;48(4):297-306
52. Caro CG, Fitz-Gerald JM, Schroter RC. Atheroma and arterial wall shear. Observation, correlation and proposal of a shear dependent mass transfer mechanism for atherogenesis. Proc Royal Soc. 1971;B.177:109–159.
53. Caro CG. Discovery of the role of wall shear in atherosclerosis. Arterioscler Thromb Vasc Biol. 2009 Feb;29(2):158-61.
54. Dietary Reference Intakes for Water, Potassium, Sodium, Chloride and Sulfate / Appendix J. Serum Electrolyte Concentrations NHANESIII. 1988-94. In: Standing Committee on the Scientific Evaluation of Dietary Reference Intakes FaNB, editor. Panel on Dietary Reference Intakes for Electrolytes and Water. Washington, DC 20001: THE NATIONAL ACADEMIES PRESS;2005. P. 558-63.
55. Oh SW, Baek SH, An JN, Goo HS, Kim S, Na KY, Chae DW, Kim S, Chin HJ. Small increases in plasma sodium are associated with higher risk of mortality in a healthy population.
J Korean Med Sci. 2013 Jul;28(7):1034-40.
56. Komiya I, Yamada T, Takasu N, Asawa T, Akamine H, Yagi N, Nagasawa Y, Ohtsuka H, Miyahara Y, Sakai H, Sato A, Aizawa T. An abnormal sodium metabolism in Japanese patients with essential hypertension, judged by serum sodium distribution, renal function and the renin-aldosterone system. J Hypertens. 1997 Jan;15(1):65-72.
57. He FJ, Markandu ND, Sagnella GA, de Wardener HE, MacGregor GA. Plasma sodium: ignored and underestimated. Hypertension. 2005 Jan;45(1):98-102.
58. Suckling RJ, He FJ, Markandu ND, MacGregor GA. Dietary salt influences postprandial plasma sodium concentration and systolic blood pressure. Kidney Int. 2012 Feb;81(4):407-11.
59. Kresser C. Shaking Up The Salt Myth: Healthy Salt Recommendations. May 4, 2012. //chriskresser.com/shaking-up-the-salt-myth-healthy-salt-recommendations/
60. Staley B, Mason H. Make it Paleo: Over 200 Grain-Free Recipes for Any Occasion. Foreword by Mark Sisson. Victory Belt Publishing, Las Vegas NV, 2011.
61. //www.marksdailyapple.com/salt-what-is-it-good-for/
62. Fragoso S. Everyday Paleo. Foreword by Robb Wolf. Victory Belt Publishing, Las Vegas NV, 2011.
63. Gower C. Paleo Slow Cooking: Gluten Free Recipes Made Simple. Foreword by Robb Wolf. Victory Belt Publishing, Las Vegas NV, 2012.
64. Mayfield J, Mayfield C. Paleo Comfort Foods: Homestyle Cooking for a Gluten-Free Kitchen. Foreword by Robb Wolf. Victory Belt Publishing, Las Vegas NV, 2011.
65. https://www.thepaleomom.com/is-salt-paleo/
66. //paleoleap.com/salt-and-a-paleo-diet/
67. //www.paleoplan.com/ingredients/salt/
68. //synergyhw.blogspot.com/2013/05/salt-one-of-your-best-friends-on-paleo.html
69. Jeggle P, Hofschröer V, Maase M, Bertog M, Kusche-Vihrog K. Aldosterone synthase knockout mouse as a model for sodium-induced endothelial sodium channel up-regulation in vascular endothelium. FASEB J. 2016 Jan;30(1):45-53.
70. Oberleithner H, Riethmüller C, Schillers H, MacGregor GA, de Wardener HE, Hausberg M. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc Natl Acad Sci U S A. 2007 Oct 9;104(41):16281-16286.
71. Ghimire K, Altmann HM, Straub A, Isenberg JS. Nitric oxide: What’s new to NO? Am J Physiol Cell Physiol. 2016 Dec 14:ajpcell.00315.2016. doi: 10.1152/ajpcell.00315.2016. [Epub ahead of print]
72. Kusche-Vihrog K, Schmitz B, Brand E. Salt controls endothelial and vascular phenotype. Pflugers Arch. 2015 Mar;467(3):499-512
73. Edwards DG, Farquhar WB. Vascular effects of dietary salt. Curr Opin Nephrol Hypertens. 2015 Jan;24(1):8-13
74. Dickinson KM, Clifton PM, Burrell LM, Barrett PH, Keogh JB. Postprandial effects of a high salt meal on serum sodium, arterial stiffness, markers of nitric oxide production and markers of endothelial function. Atherosclerosis. 2014 Jan;232(1):211-6.
75. Blanch N, Clifton PM, Petersen KS, Keogh JB.Effect of sodium and potassium supplementation on vascular and endothelial function: a randomized controlled trial. Am J Clin Nutr. 2015 May;101(5):939-46
76. Wild J, Soehnlein O, Dietel B, Urschel K, Garlichs CD, Cicha I. Rubbing salt into wounded endothelium: sodium potentiates proatherogenic effects of TNF-α under non-uniform shear stress. Thromb Haemost. 2014 Jul 3;112(1):183-95.
77. Dmitrieva NI, Burg MB. Elevated sodium and dehydration stimulate inflammatory signaling in endothelial cells and promote atherosclerosis. PLoS One. 2015 Jun 4;10(6):e0128870. doi: 10.1371/journal.pone.0128870.
78. Oberleithner H, Callies C, Kusche-Vihrog K, Schillers H, Shahin V, Riethmüller C, Macgregor GA, de Wardener HE. Potassium softens vascular endothelium and increases nitric oxide release. Proc Natl Acad Sci U S A. 2009 Feb 24;106(8):2829-34
Loren Cordain, Ph.D.
As a professor at Colorado State University, Dr. Loren Cordain developed The Paleo Diet® through decades of research and collaboration with fellow scientists around the world.
More About The Author
Health Benefits and Prevention





The Paleo Diet® Newsletter
Sign up to get info on healthy nutrition, free recipes, meal prepping, and the latest health research. New subscribers will also receive our Official Paleo Grocery List!