Wheat Series Part 5: How Wheat Can Trigger Chronic Disease

Russian roulette is a game that few ever want to play. A revolver is loaded with a single bullet. The players take turns putting the gun to their heads and pulling the trigger. Eventually someone gets a loaded chamber and pays the ultimate price.
We’ve known for a while that most chronic diseases such as cancer, autoimmune disease, and heart disease have a genetic component.1-10 Genetics are the loaded bullet that we sadly have no control over.
For celiac disease, the “bullet” is a genetic variant in HLA-DQ.11, 12 However, a large number of people with the variant never express the disease. Further, those who do develop the condition usually resolve it by removing gluten from their diet.13, 14 In other words, the bullet might be in the chamber, but often the gun is never fired. Environmental factors ultimately pull the trigger.
In the first four parts of this series we talked about how wheat (and to a degree other gluten-containing grains such as rye and barley) is highly effective at dysregulating the immune system of our guts. In fact, it’s the only food we know of that affects all three pathways of dysregulation:
- Opening up the tight junctions of our gut (Part 2)
- Excess and chronic bacterial stress (Part 3)
- Harmful dietary antigens (Part 4)
In this final part, we’ll talk about how the resulting chronic inflammation leads to a pathological state that can essentially trigger chronic disease. But just as importantly, we’ll discuss how there has to be a bullet in the chamber first. The genetic susceptibility has to be there.
Building Inflammation
Our genetics have not changed in the last 100 years. Yet, chronic disease such as autoimmune conditions and cancer have risen dramatically—faster than rate of population growth.
In other words (going with our analogy), the number of bullets hasn’t changed, but for some reason the trigger is getting pulled a lot more often. Which is surprising considering no one wants to pull it.
From what little we understand, Russian roulette players essentially have to build up to it, engaging in other risky behavior and slowly desensitizing themselves.15 Likewise, our bodies have a lot of defenses to avoid ever pulling the trigger on disease, even when the bullet is there.
So, while we hopefully made the case in the previous four parts on why wheat is not good for us, one piece of bread isn’t going to give you cancer. Despite all the dysregulation of our immune system caused by wheat, it still takes a lot to build up to the point of disease.16, 17
In fact, as we discussed in Part 1, all the inflammatory processes activated by wheat are both normal and necessary actions designed to deal with regular bacterial stress. Mice bred without these inflammatory responses suffer severe tissue damage and wasting disease.18, 19
Even the temporary shift in the balance between two critical immune cells—Treg and Th17 cells—is a natural response to this inflammation. Let’s explore these two cells a little more.
Th17 Cells and Inflammation
In a healthy state, Treg cells dominate. Their role is to suppress the immune system,20-24 preventing it from damaging our own bodies. People unfortunate enough to have dysfunctional Treg cells suffer severe autoimmune disease.25
Th17, on the other hand, has a murkier and less benign role. Only discovered in 2006, it solved an important puzzle for researchers. Scientists knew that T cells were involved in many conditions but none of the known T cells at the time fully explained disease development.26 With the discovery of Th17, they had their answer.26, 27
Proving to be highly inflammatory cells, they effectively explained the damage in a multitude of chronic diseases16, 28-31 such as asthma,32 heart disease,33, 34 and most autoimmune conditions30, 35 including celiac disease,36, 37 type 1 diabetes,38, 39 Crohn’s disease,40, 41 rheumatoid arthritis,31, 42 and multiple sclerosis.43
A question remained, however: Why would our bodies produce such a self-destructive cell?
The reason lies in their role. It was believed that Th17 cells evolved to deal with harmful bacterial infections, and effectively handling an invasion means doing some damage to our own bodies.19, 44, 45
This damage seems to be acceptable to our bodies and even part of a healthy immune response as long as one essential condition is met: the shift toward Th17 dominance is short-lived and ends. Once an infection is dealt with and the resulting inflammation quiets down, Th17 cells die off and Treg cells return to dominating our immune system.24, 46
So what happens if the inflammation doesn’t end?
According to one emerging theory, the result is an out-of-control pathogenic form of the Th17 cell.10, 23, 24 In other words, if normal bacterial stress causes a little risky behavior by inciting beneficial Th17, chronic inflammation causes the buildup that leads to the pathogenic Th17 triggering chronic disease.
How Chronic Inflammation Occurs
The diagram below shows the different responses between normal and chronic inflammation.30
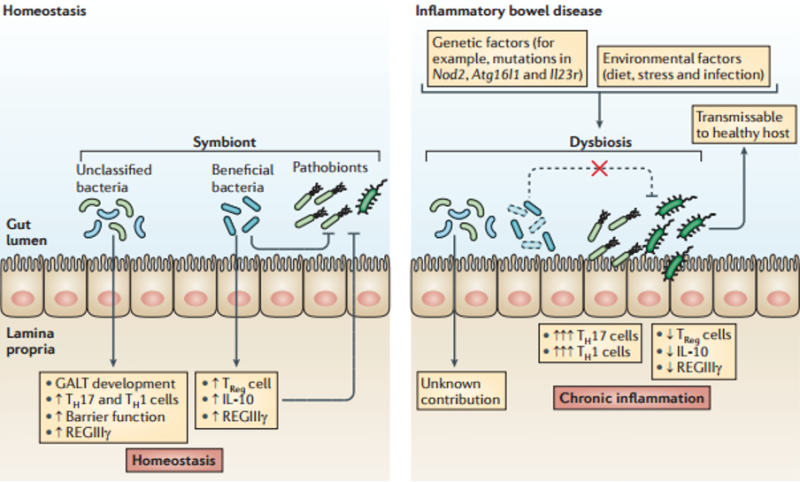
Let’s explore this destructive shift a little more closely. Under normal inflammation, the number of Treg cells increase alongside the Th17 cells, allowing Treg cells to continue controlling Th17’s destructive potential and maintain some balance.30, 47
But in chronic inflammation, a dramatic shift occurs. More and more innate immune cells such as dendritic cells and CD14+ macrophages (immune cells explained in Part 3) are activated or recruited to the digestive immune system.17, 28, 48, 49
Over time, these cells change the chemical milieu of the gut to one that is highly inflammatory. IL-23 is released which both promotes the destructive form of Th17 and inhibits Treg.27, 47, 50, 51 Newly recruited CD14+ macrophages also suppress Treg.52
In fact, the chronic inflammation causes Treg cells to “flip” and start behaving like Th17, contributing to the inflammation instead of preventing it.47, 52, 53
The result is that chronic inflammation breaks the Treg/Th17 balance. Treg cells lose their ability to control the immune system and Th17 cells, now uninhibited, take on a more destructive form, traveling from the gut45 throughout the body.30
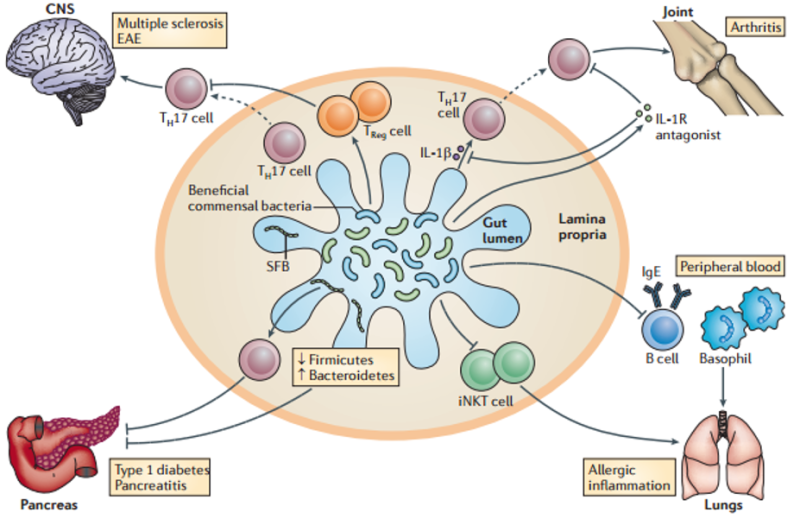
So, where does wheat come into all of this?
Wheat, as we’ve shown previously, creates the chronic inflammation that sets off this cascade. In fact, in one study of mice that tested many dietary antigens, wheat was the only one that could activate inflammatory Th17 cells.54
Put simply, wheat sets in motion the build-up that causes our bodies to ultimately trigger chronic disease.
Why Doesn’t Wheat Trigger Chronic Disease in Everyone?
The very sobering thought is that the chronic inflammation, which wheat is so effective at creating (in fact it took three parts to explain all the ways wheat can cause it), appears to be common to everyone.17, 20, 55 So why aren’t we all sick?
Remember there are two parts to the Russian roulette analogy. Wheat causes our immune system to put the gun to our heads and pull the trigger, but there still needs to be a bullet in the chamber. The genetic susceptibility has to be there.
Sure enough, a genetic susceptibility to chronic inflammation has been identified in many conditions. Often taking the form of a hyper-sensitivity to inflammation or a failure of the Treg system to suppress it.
CD14+ macrophages appear to be particularly potent in rheumatoid arthritis.56 Celiacs are hyper-sensitive to IL-15—one of the key proteins used by wheat to produce inflammation.3 Much higher levels of inflammatory CD14+ macrophages exist in the guts of people with inflammatory bowel disease (IBD).57 IBD sufferers also appear to be more responsive to IL-23.29 In type 2 diabetes, the immune cells that destroy the pancreas exist in healthy and afflicted subjects, but Treg cells appear to be less functional in diabetics.1, 2
Notably, people with one of these conditions are often more susceptible to the others.58-63
Still, There Are a Lot of Bullets
The need for a “genetic bullet” in order for a disease to materialize has led many to breathe a sigh of relief. An example is this 2015 Washington Post article: “For many, gluten isn’t the villain it gets cracked up to be.” But the fact is that there are many chronic diseases, and they are all on the rise.
Recent research is showing more and more that inappropriate chronic inflammation is at the heart of almost every “disease of civilization” including cancer,64, 65 metabolic disorders,66, 67 Alzheimer’s disease,68 most autoimmune conditions,30, 35 and heart disease where aberrant macrophages form the atherosclerotic plaques.69, 70 That amounts to a whole lot of genetic bullets.
While the research is still small, several of these conditions—including celiac disease, diabetes, and IBD—are improved when wheat is removed from the diet.13, 14, 71 – 73
So feel free to do as the Washington Post article says: eat your bread, and trust your luck that the chamber is empty. But with that many potential bullets in the revolver and chronic inflammation (so effectively produced by wheat) ready to pull the trigger, I’m personally going to avoid it.
Explore the rest of our Wheat Series:
Part 1: The Digestive Immune System
Part 2: Wheat and Gluten’s Effect on Intestinal Permeability
Part 3: How Wheat Mimics Bacteria
Part 4: Wheat as a Harmful Dietary Antigen
References
- Danke, N.A., et al., Comparative study of GAD65-specific CD4+ T cells in healthy and type 1 diabetic subjects. J Autoimmun, 2005. 25(4): p. 303-11.
- Richer, M.J., et al., Immunomodulation of antigen presenting cells promotes natural regulatory T cells that prevent autoimmune diabetes in NOD mice. PLoS One, 2012. 7(2): p. e31153.
- Harris, K.M., A. Fasano, and D.L. Mann, Monocytes differentiated with IL-15 support Th17 and Th1 responses to wheat gliadin: implications for celiac disease. Clin Immunol, 2010. 135(3): p. 430-9.
- Cereijido, M., et al., New diseases derived or associated with the tight junction. Arch Med Res, 2007. 38(5): p. 465-78.
- Fasano, A., Physiological, Pathological, and Therapeutic Implications of Zonulin-Mediated Intestinal Barrier Modulation Living Life on the Edge of the Wall. American Journal of Pathology, 2008. 173(5): p. 1243-1252.
- Fasano, A., Zonulin and its regulation of intestinal barrier function: the biological door to inflammation, autoimmunity, and cancer. Physiol Rev, 2011. 91(1): p. 151-75.
- Ahlbom, A., et al., Cancer in twins: Genetic and nongenetic familial risk factors. Journal of the National Cancer Institute, 1997. 89(4): p. 287-293.
- Marenberg, M.E., et al., GENETIC SUSCEPTIBILITY TO DEATH FROM CORONARY HEART-DISEASE IN A STUDY OF TWINS. New England Journal of Medicine, 1994. 330(15): p. 1041-1046.
- Heward, J. and S.C.L. Gough, Genetic susceptibility to the development of autoimmune disease. Clinical Science, 1997. 93(6): p. 479-491.
- Levin, L. and Y. Tomer, The etiology of autoimmune diabetes and thyroiditis: evidence for common genetic susceptibility. Autoimmunity Reviews, 2003. 2(6): p. 377-386.
- Sollid, L.M., et al., EVIDENCE FOR A PRIMARY ASSOCIATION OF CELIAC-DISEASE TO A PARTICULAR HLA-DQ ALPHA-BETA HETERODIMER. Journal of Experimental Medicine, 1989. 169(1): p. 345-350.
- Sollid, L.M. and E. Thorsby, HLA SUSCEPTIBILITY GENES IN CELIAC-DISEASE – GENETIC-MAPPING AND ROLE IN PATHOGENESIS. Gastroenterology, 1993. 105(3): p. 910-922.
- Johnston, S.D., C. Rodgers, and R.G.P. Watson, Quality of life in screen-detected and typical coeliac disease and the effect of excluding dietary gluten. European Journal of Gastroenterology & Hepatology, 2004. 16(12): p. 1281-1286.
- Fasano, A. and C. Catassi, Current approaches to diagnosis and treatment of celiac disease: An evolving spectrum. Gastroenterology, 2001. 120(3): p. 636-651.
- Collins, K.A., Adolescent Russian roulette deaths. Am J Forensic Med Pathol, 2010. 31(1): p. 4-6.
- Gonzalez-Quintial, R., et al., Systemic autoimmunity and lymphoproliferation are associated with excess IL-7 and inhibited by IL-7Ralpha blockade. PLoS One, 2011. 6(11): p. e27528.
- Palova-Jelinkova, L., et al., Gliadin fragments induce phenotypic and functional maturation of human dendritic cells. J Immunol, 2005. 175(10): p. 7038-45.
- Muramatsu, M., et al., Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell, 2000. 102(5): p. 553-63.
- Reynolds, J.M., et al., Cutting edge: regulation of intestinal inflammation and barrier function by IL-17C. J Immunol, 2012. 189(9): p. 4226-30.
- du Pre, M.F. and J.N. Samsom, Adaptive T-cell responses regulating oral tolerance to protein antigen. Allergy, 2011. 66(4): p. 478-90.
- Battaglia, M., et al., IL-10-producing T regulatory type 1 cells and oral tolerance. Ann N Y Acad Sci, 2004. 1029: p. 142-53.
- Wing, K. and S. Sakaguchi, Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol, 2010. 11(1): p. 7-13.
- Williamson, E., G.M. Westrich, and J.L. Viney, Modulating dendritic cells to optimize mucosal immunization protocols. J Immunol, 1999. 163(7): p. 3668-75.
- Veldman, C., A. Nagel, and M. Hertl, Type I regulatory T cells in autoimmunity and inflammatory diseases. International Archives of Allergy and Immunology, 2006. 140(2): p. 174-183.
- Scalapino, K.J. and D.I. Daikh, CTLA-4: a key regulatory point in the control of autoimmune disease. Immunol Rev, 2008. 223: p. 143-55.
- Mesquita Jr, D., et al., Autoimmune diseases in the TH17 era. Braz J Med Biol Res, 2009. 42(6): p. 476-86.
- Langrish, C.L., et al., IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med, 2005. 201(2): p. 233-40.
- Smith, P.D., et al., Intestinal macrophages and response to microbial encroachment. Mucosal Immunol, 2011. 4(1): p. 31-42.
- Ohnmacht, C., et al., Intestinal microbiota, evolution of the immune system and the bad reputation of pro-inflammatory immunity. Cell Microbiol, 2011. 13(5): p. 653-9.
- Kamada, N., et al., Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol, 2013. 13(5): p. 321-35.
- Tesmer, L.A., et al., Th17 cells in human disease. Immunological Reviews, 2008. 223: p. 87-113.
- Cosmi, L., et al., Th17 cells: new players in asthma pathogenesis. Allergy, 2011. 66(8): p. 989-98.
- Taleb, S., A. Tedgui, and Z. Mallat, IL-17 and Th17 cells in atherosclerosis: subtle and contextual roles. Arterioscler Thromb Vasc Biol, 2015. 35(2): p. 258-64.
- van Bruggen, N. and W. Ouyang, Th17 cells at the crossroads of autoimmunity, inflammation, and atherosclerosis. Immunity, 2014. 40(1): p. 10-2.
- Singh, R.P., et al., Th17 cells in inflammation and autoimmunity. Autoimmun Rev, 2014. 13(12): p. 1174-81.
- Monteleone, I., et al., Characterization of IL-17A-producing cells in celiac disease mucosa. J Immunol, 2010. 184(4): p. 2211-8.
- Castellanos-Rubio, A., et al., TH17 (and TH1) signatures of intestinal biopsies of CD patients in response to gliadin. Autoimmunity, 2009. 42(1): p. 69-73.
- Kumar, P. and G. Subramaniyam, Molecular underpinnings of Th17 immune-regulation and their implications in autoimmune diabetes. Cytokine, 2015. 71(2): p. 366-76.
- Shao, S., et al., Th17 cells in type 1 diabetes. Cell Immunol, 2012. 280(1): p. 16-21.
- Elson, C.O., et al., Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology, 2007. 132(7): p. 2359-70.
- Brand, S., Crohn’s disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut, 2009. 58(8): p. 1152-67.
- Hirota, K., et al., Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med, 2007. 204(12): p. 2803-12.
- Du, C., et al., MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol, 2009. 10(12): p. 1252-9.
- McFall-Ngai, M., Adaptive immunity: care for the community. Nature, 2007. 445(7124): p. 153.
- Ivanov, II, et al., Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell, 2009. 139(3): p. 485-98.
- Grossman, Z., et al., Concomitant regulation of T-cell activation and homeostasis. Nat Rev Immunol, 2004. 4(5): p. 387-95.
- Torchinsky, M.B., et al., Innate immune recognition of infected apoptotic cells directs T(H)17 cell differentiation. Nature, 2009. 458(7234): p. 78-82.
- Nikulina, M., et al., Wheat gluten causes dendritic cell maturation and chemokine secretion. J Immunol, 2004. 173(3): p. 1925-33.
- Yamazaki, K., J.A. Murray, and H. Kita, Innate immunomodulatory effects of cereal grains through induction of IL-10. Journal of Allergy and Clinical Immunology, 2008. 121(1): p. 172-178.
- da Silva Martins, M. and C.A. Piccirillo, Functional stability of Foxp3+ regulatory T cells. Trends Mol Med, 2012. 18(8): p. 454-62.
- Lochner, M., et al., In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORgamma t+ T cells. J Exp Med, 2008. 205(6): p. 1381-93.
- Evans, H.G., et al., Optimal induction of T helper 17 cells in humans requires T cell receptor ligation in the context of Toll-like receptor-activated monocytes. Proc Natl Acad Sci U S A, 2007. 104(43): p. 17034-9.
- Voo, K.S., et al., Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci U S A, 2009. 106(12): p. 4793-8.
- Shi, G., et al., Cell-cell interaction with APC, not IL-23, is required for naive CD4 cells to acquire pathogenicity during Th17 lineage commitment. J Immunol, 2012. 189(3): p. 1220-7.
- Bernardo, D., et al., Is gliadin really safe for non-coeliac individuals? Production of interleukin 15 in biopsy culture from non-coeliac individuals challenged with gliadin peptides. Gut, 2007. 56(6): p. 889-890.
- Evans, H.G., et al., In vivo activated monocytes from the site of inflammation in humans specifically promote Th17 responses. Proc Natl Acad Sci U S A, 2009. 106(15): p. 6232-7.
- Kamada, N., et al., Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest, 2008. 118(6): p. 2269-80.
- Weng, X., et al., Clustering of inflammatory bowel disease with immune mediated diseases among members of a northern california-managed care organization. Am J Gastroenterol, 2007. 102(7): p. 1429-35.
- Holmes, G.K.T., Coeliac disease and Type 1 diabetes mellitus – the case for screening. Diabetic Medicine, 2001. 18(3): p. 169-177.
- Not, T., et al., Undiagnosed coeliac disease and risk of autoimmune disorders in subjects with Type I diabetes mellitus. Diabetologia, 2001. 44(2): p. 151-155.
- Valerio, G., et al., Severe clinical onset of diabetes and increased prevalence of other autoimmune diseases in children with coeliac disease diagnosed before diabetes mellitus. Diabetologia, 2002. 45(12): p. 1719-1722.
- Pohjankoski, H., et al., Diabetes, coeliac disease, multiple sclerosis and chronic arthritis in first-degree relatives of patients with juvenile idiopathic arthritis. Acta Paediatrica, 2012. 101(7): p. 767-771.
- Binus, A.M., et al., Associated comorbidities in psoriasis and inflammatory bowel disease. Journal of the European Academy of Dermatology and Venereology, 2012. 26(5): p. 644-650.
- Coussens, L.M. and Z. Werb, Inflammation and cancer. Nature, 2002. 420(6917): p. 860-867.
- Grivennikov, S.I., F.R. Greten, and M. Karin, Immunity, Inflammation, and Cancer. Cell, 2010. 140(6): p. 883-899.
- Hotamisligil, G.S., Inflammation and metabolic disorders. Nature, 2006. 444(7121): p. 860-867.
- Festa, A., et al., Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS). Circulation, 2000. 102(1): p. 42-7.
- Akiyama, H., et al., Inflammation and Alzheimer’s disease. Neurobiology of Aging, 2000. 21(3): p. 383-421.
- Libby, P., P.M. Ridker, and A. Maseri, Inflammation and atherosclerosis. Circulation, 2002. 105(9): p. 1135-1143.
- Linton, M.F. and S. Fazio, Macrophages, inflammation, and atherosclerosis. International Journal of Obesity, 2003. 27: p. S35-S40.
- Antvorskov, J.C., et al., Dietary gluten and the development of type 1 diabetes. Diabetologia, 2014. 57(9): p. 1770-1780.
- Sildorf, S.M., et al., Remission without insulin therapy on gluten-free diet in a 6-year old boy with type 1 diabetes mellitus. BMJ Case Rep, 2012. 2012.
- Shahbazkhani, B., et al., Non-Celiac Gluten Sensitivity Has Narrowed the Spectrum of Irritable Bowel Syndrome: A Double-Blind Randomized Placebo-Controlled Trial. Nutrients, 2015. 7(6): p. 4542-4554.
Food Sensitivities





The Paleo Diet® Newsletter
Sign up to get info on healthy nutrition, free recipes, meal prepping, and the latest health research. New subscribers will also receive our Official Paleo Grocery List!